What is Cystic Fibrosis?
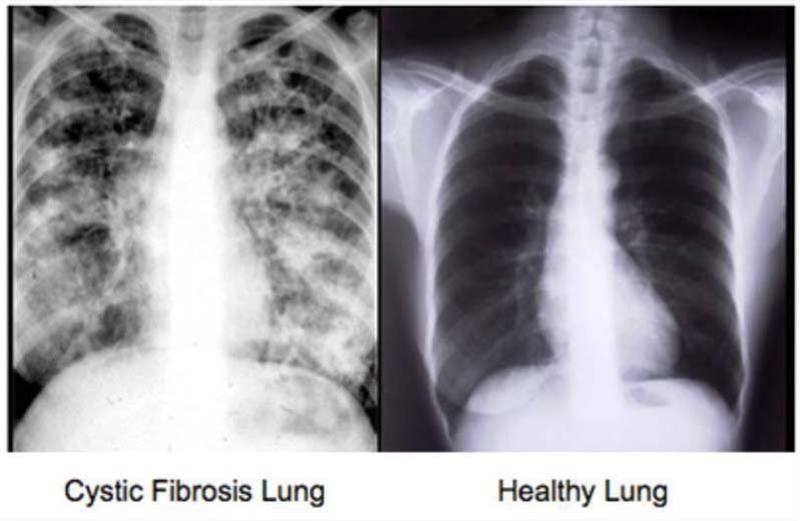
Cystic fibrosis is an inherited condition in which the lungs and digestive system can become clogged with thick, sticky mucus.
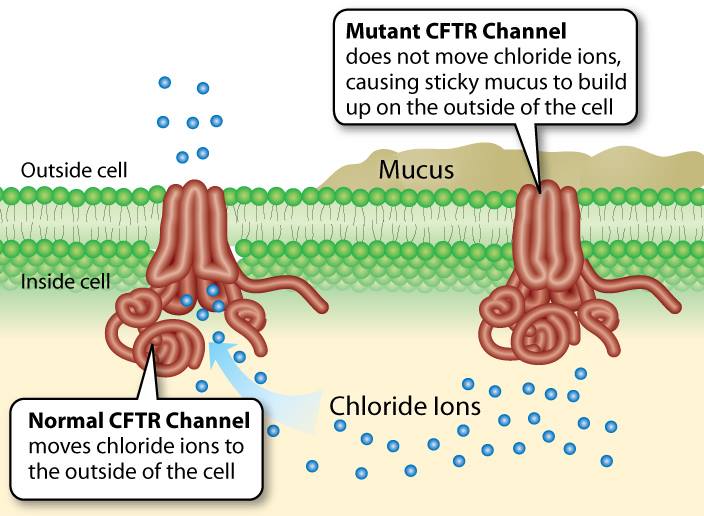
It is an inherited autosomal disorder that affects the ability of epithelial cells of the body to effectively transport chloride.
Secondary to defective chloride transport, water transport is also deregulated, and many organs, including the lungs, pancreas, and liver, are adversely affected.
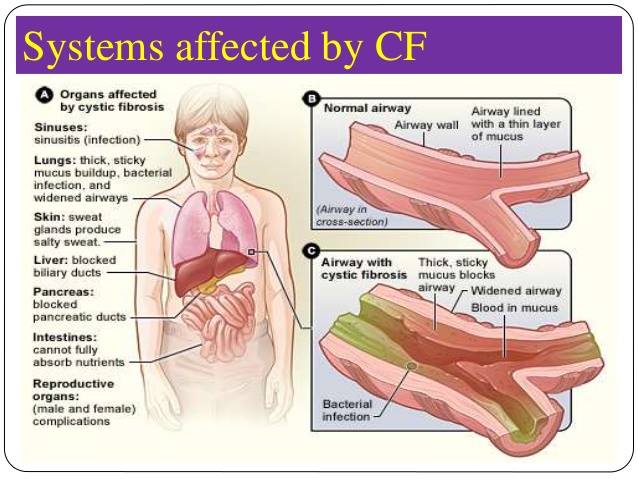
In the lungs, mucous secretions accumulate and are unable to be cleared from the airways. The mucous buildup eventually leads to airway obstruction, predisposing patients with CF to infection. These infections are more frequent, more severe.
Cystic fibrosis (CF) is a genetic condition affecting more than 10,800 people in the UK. You are born with CF and cannot catch it later in life, but one in 25 of us carry the faulty gene that causes it, usually without knowing.
Cystic fibrosis and infection
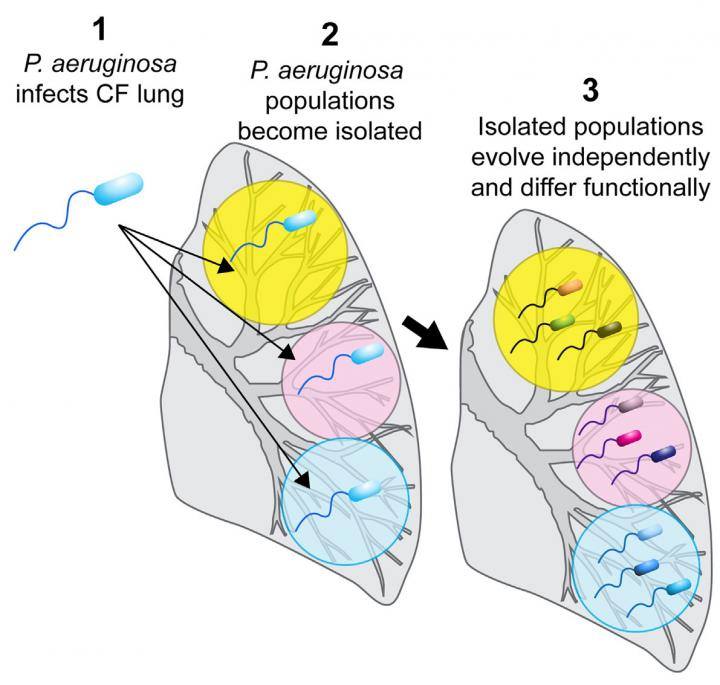
Bacteria commonly isolated from CF sputum include Staphylococcus aureus, Haemophilus influenza and Pseudomonas aeruginosa. Colonization of the airways by mucoid, alginate-producing variants of P. aeruginosa is recognized as a major cause of pulmonary deterioration
Genetics & diagnosis
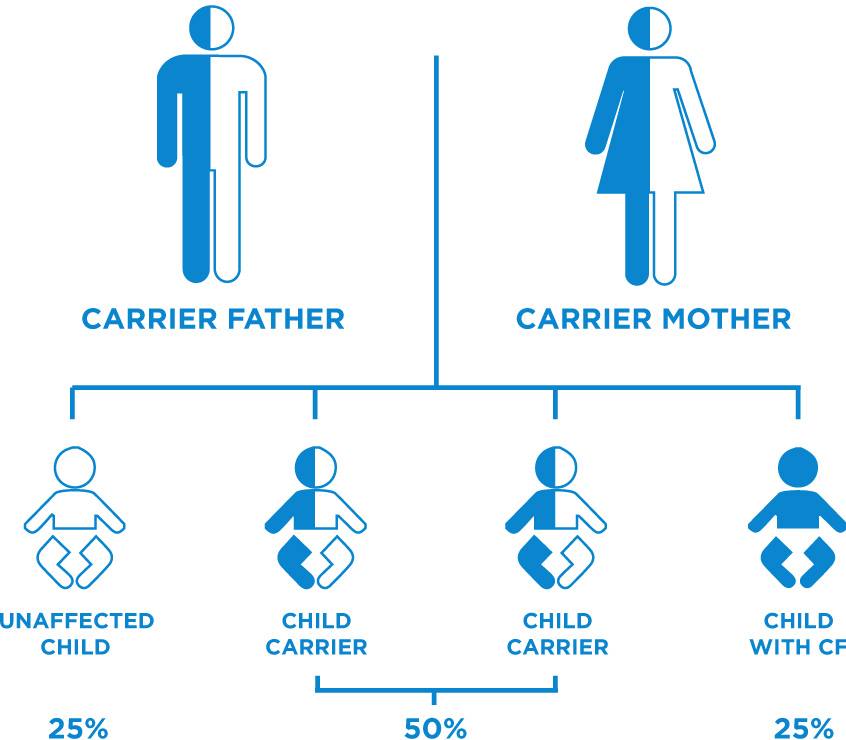
To have CF, you need to have inherited two faulty copies of the gene (one from each parent), Their parents will not usually have the condition themselves, because they will only carry one faulty gene and one that works normally. While people with CF often look healthy on the outside, each individual is battling their own range of symptoms on a daily basis
Effect on lung
If a bad lung infection occurs, which can happen often in some individuals, intravenous antibiotics are required, which may have to be administered in hospital.
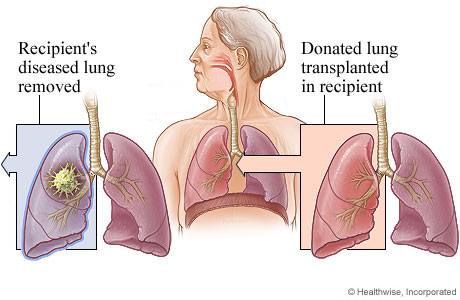
For many people with CF a double lung transplant becomes the only option. While this tends to improve quality of life, the operation comes with risks and after a transplant certain drugs are required to prevent the donor organs from being rejected.
Effect on Digestive system
• Ongoing diarrhea or bulky, foul-smelling, greasy stools.
• Intestinal blockages, especially in newborns.
• Poor weight gain and growth in children because they are unable to absorb fats and proteins.
• Pancreatitis.
• Liver disease.
• Diabetes.
• Gallstones
Effect on Bones
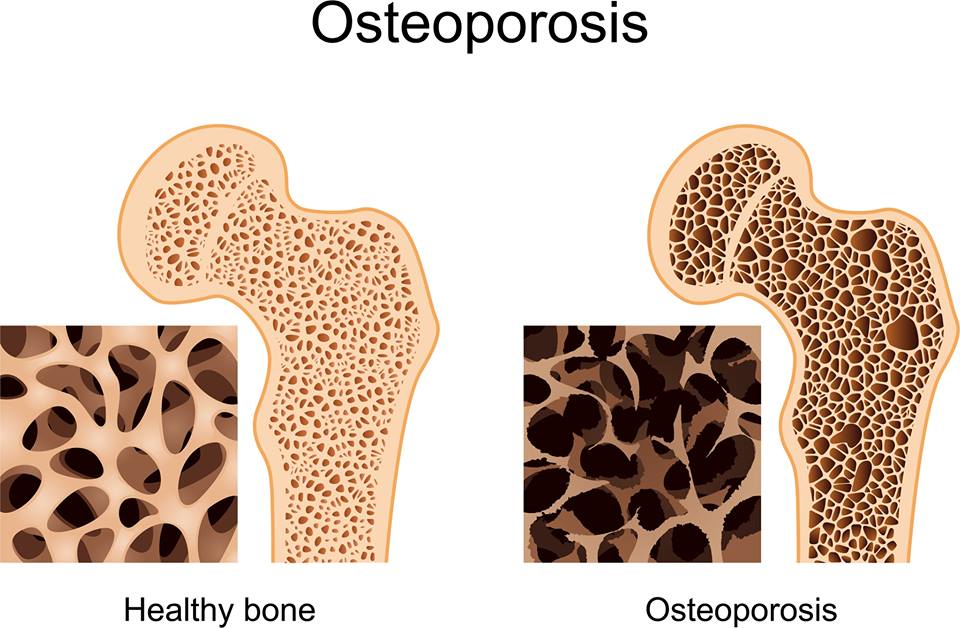
A lack of minerals caused by a damaged pancreas can result in osteoporosis (weak or brittle bones). Bisphosphonates, which are used to treat osteoporosis in post-menopausal women, have been shown to help treat osteoporosis in cystic fibrosis. Researchers are examining the benefits of high doses of vitamin D and calcium.
Symptoms of cystic fibrosis
Symptoms of cystic fibrosis tend to start in early childhood, although they can sometimes develop very soon after birth, or may not be obvious until adulthood.
Additional complications
Distal intestinal obstruction syndrome (DIOS)
Previously known as meconium ileus equivalent (MIE), DIOS is a condition that is unique to cystic fibrosis. DIOS causes blockages in the stomach, causing symptoms such as colicky stomach pain, bloating, nausea and weight loss. In most cases DIOS can be controlled with medication.
Adequate hydration and an aggressive regimen of laxatives are essential for treatment and prevention of DIOS. Osmotic laxatives such as polyethylene glycol are preferred.
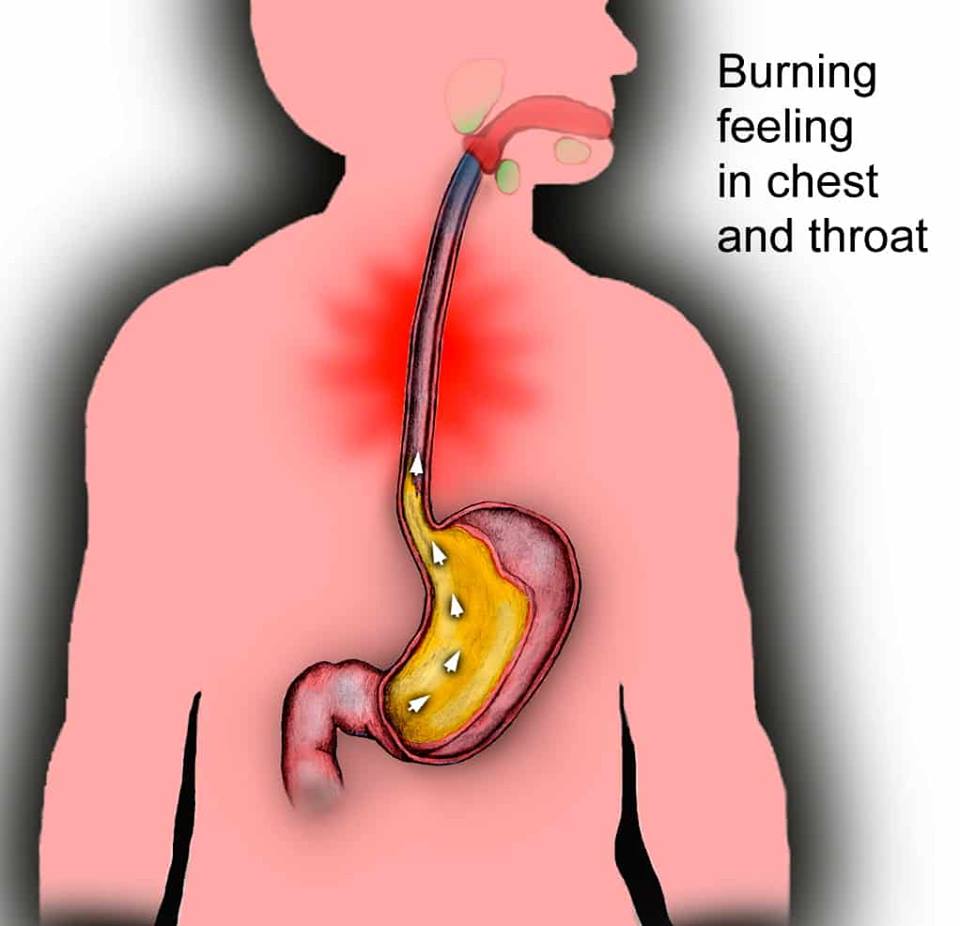
Gastro-esophageal reflux disease (GORD)
A common occurrence in people with CF. Symptoms includes heartburn, a nasty sour taste in the mouth, and pain or difficulty swallowing. This can be treated with antacids in most cases.
Screening and testing for cystic fibrosis
If you have a family history of cystic fibrosis, you can be tested to determine if you're at risk of having a child with the condition by checking if you're a "carrier" of the faulty gene that causes it.
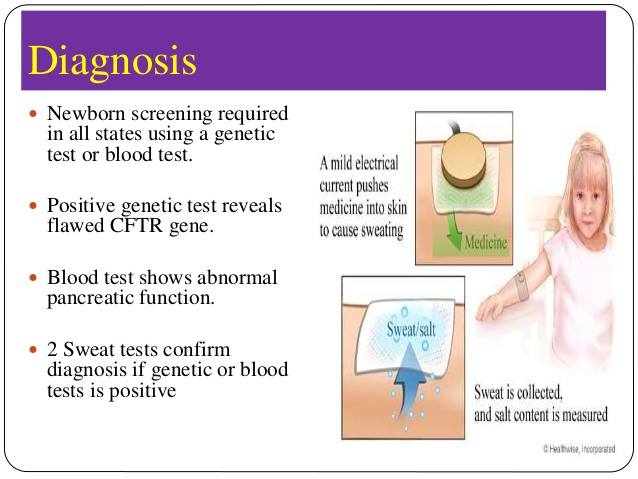
Most cases of cystic fibrosis are now detected soon after birth through the newborn blood spot test.
This involves collecting a drop of blood from the baby's heel and testing it for abnormalities that could indicate cystic fibrosis.
More tests will be needed to confirm the diagnosis, such as:
• A sweat test – to measure the amount of salt in sweat, as the sweat of someone with cystic fibrosis has higher levels of salt than normal.
• A genetic test – where a sample of blood or saliva is checked for the faulty gene that causes cystic fibrosis.